�l(f��)���r(sh��)�g��2022-04-09���ٷ���r(n��ng)�I(y��)Փ���g�[��1��
ժ Ҫ�� ժҪ �S���y(c��)���g(sh��)�IJ���l(f��)չ��Խ��(l��i)Խ����N��ȫ����M��(sh��)��(j��)���y(c��)���͏V����(y��ng)�á��ڶ�������M��(sh��)��(j��)���l(f��)ʽ���L(zh��ng)��ͬ�r(sh��)�����˺˻���M��(sh��)��(j��)�������w����M��(sh��)��(j��)Ҳ�dz���Ҫ����ͨ���y(c��)���ȫ����M�����г��˺˻���M����Ҳ���������w����M���У���Ώĺ�����ȫ����M��(sh��)��(j��)����ȡ
����ժҪ �S���y(c��)���g(sh��)�IJ���l(f��)չ��Խ��(l��i)Խ����N��ȫ����M��(sh��)��(j��)���y(c��)���͏V����(y��ng)�á��ڶ�������M��(sh��)��(j��)���l(f��)ʽ���L(zh��ng)��ͬ�r(sh��)�����˺˻���M��(sh��)��(j��)�������w����M��(sh��)��(j��)Ҳ�dz���Ҫ����ͨ���y(c��)���ȫ����M�����г��˺˻���M����Ҳ���������w����M���У���Ώĺ�����ȫ����M��(sh��)��(j��)����ȡ��ƴ�b�����w����M���в����ԑ�(y��ng)�óɞ龀���w����M�ڷ�������W(xu��)���z���W(xu��)���t(y��)�W(xu��)�ȷ�����о�����֮һ�����ڴˣ���ȫ����M��(sh��)��(j��)����ȡ�����w����M���еIJ��Լ����P(gu��n)��ܛ������l(f��)չ������(j��)��ȫ����M��(sh��)��(j��)���^�������w reads �ķ�ʽ�ͺ��m(x��)ƴ�b���ԵIJ�ͬ�����Է֞��Ѕ�������ƴ�b�����͏��^ƴ�b��������ͬƴ�b���Լ�ܛ��Ҳ���F(xi��n)�����Եă�(y��u)��(sh��)�;����ԡ����Ŀ��Y(ji��)�����^�ˮ�(d��ng)ǰ��ȫ����M��(sh��)��(j��)�Ы@�þ����w����M��(sh��)��(j��)�IJ��Ժ�ܛ����(y��ng)�ã�����(du��)ʹ������ʹ�ò�ͬ���Ժ����P(gu��n)ܛ������o�轨�h�����ڞ龀���w����M�������ƌW(xu��)�����P(gu��n)�о����ṩ�����ϵą�����
�����P(gu��n)�I�~ ȫ����M;�����w����M;�Ѕ�������ƴ�b����;���^ƴ�b����;ƴ�bܛ��
���������w����M(mitochondrial genome)����һ�N���������@ȡ���z����(bi��o)ӛ������и�ͻ׃���ʡ��o(w��)�����ؽM���߿�ؐ��(sh��)��ĸϵ�z�������c(di��n)[1]�����V����(y��ng)����ϵ�y(t��ng)��������������о�[2~5]��Ⱥ�w�z��[6~13]���t(y��)�W(xu��)[14~17]�����B(t��i)�W(xu��)�о�[18~20]���I(l��ng)�������ڵ��о��A�Σ������w����M���еī@ȡ������ͨ�^(gu��)�L(zh��ng)��?zh��n)����?y��ng)(long range PCR, LR-PCR)�Ϳ�¡ PCR �U(ku��)����Ȼ����ͨ�^(gu��)���ﲽ��(primer walking)ɣ��(Sanger) �y(c��)���@�N������(zh��n)�_�Ըߣ���ͨ���͡��ĕr(sh��)�����ͻ��M(f��i)�ߡ��S���y(c��)���g(sh��)�ą�չ���e����һ���y(c��)���g(sh��)(next-generation sequencing, NGS)�ą�չ���y(c��)��ɱ��Ŀ����½���ʹ�þ����w����M���еī@ȡ׃�ø������ס�Ŀǰ��NGS �����������g(sh��)(�� LRPCR �� NGS��RNA �y(c��)���ȱ�����a(b��)(gap filling)��ֱ���B�����y(c��)��[21~23]��)ʹ�ø�ͨ���y(c��)��ɞ��ձ�F(xi��n)����Ȃ��y(t��ng)�� Sanger �y(c��)���g(sh��)��NGS ���g(sh��)ͨ���ߡ����Ը��������ø��͵Ļ��M(f��i)�@��ȫ����M����(wholegenome sequencing, WGS)�����@�����кͻ����D(zhu��n)䛱�[24]����һ���y(c��)���g(sh��)�Ļ���ԭ���ǣ��y(c��)��ƽ�_(t��i)��(du��)�ӱ��� DNA ����x������ľ����w DNA �S�C(j��)���� 50~700 bp �Ć�� DNA �Ď�(k��)(DNA �L(zh��ng)��ȡ�Q���Ď�(k��)��(g��u)��ƽ�_(t��i))���Ԍ���Ƭ�εăɶ��c�y(c��)����^�����B������(l��i)��Ȼ��(du��)�a(ch��n)���Ďװ��f(w��n)�l�� DNA ����މ�Мy(c��)��Ч����(zh��n)�_�����ٵث@�ô��� DNA ���У����ͨ�^(gu��)������Ϣ�����ĺ�����ȫ����M��(sh��)��(j��)�Ы@ȡ�����w����M�������(l��i)���� Pacific Biosciences (PacBio) �� Oxford Nanopore �η��Ӝy(c��)���g(sh��)������ĵ������y(c��)���g(sh��)�w�م�չ����y(c��)���^(gu��)�̟o(w��)��މ�� DNA �S�C(j��)����� PCR �U(ku��)���������x�L(zh��ng)���ӵ���ʮ kb�������� 100 kb��ƴ�b��õ������|(zh��)����ȫ����M���С�����M���g(sh��)�ą�չҲ��ʹ�����w���Д�(sh��)��(j��)����ʽ�����ӡ���ˣ�Խ��(l��i)Խ����о��߇Lԇ���ö���(g��)��ͬ�IJ��ԏ� WGS ��(sh��)��(j��)�Ы@ȡ�����w����M[23,25~39]��
������ NGS �r(sh��)����θ�Ч���x���������w DNA ������� DNA ����Ⱦ�Ǿ����w����M�y(c��)���m(x��)�������P(gu��n)�I��Ŀǰ��Ҫ�����ɷN���x���ԣ�(1)�� NGS �y(c��)��ǰ���Ŀ� DNA ���������x���������w DNA���@�N������ͨ�^(gu��)�Ȼ��C�ܶ��ݶ��x��/�����x�Ļ���ԇ���и������錢�� DNA�;����w DNA���x[40,41]��Ȼ���x������ľ����w DNA މ���Ď�(k��)��(g��u)����ͨ���y(c��)���@�ӣ�ͨ�^(gu��)�� NGS �y(c��)��ǰ�͌��� DNA �;����w DNA (���~�G�w DNA)���x���Ա��C�@�õĔ�(sh��)��(j��)�ǁ�(l��i)���ھ����w(���~�G�w)��ԓ�����ă�(y��u)��(sh��)���ڱ����˺� DNA ����Ⱦ���������w�����D(zhu��n)�Ƶ��˻��������(nuclear mitochondrial pseudogenes, Numts[42])�����ǣ��������x�����ķ������õ�ԇ���Ѓr(ji��)���F���������^�����ͺĕr(sh��)��������(du��)��Ʒ���|(zh��)���͔�(sh��)��Ҳ����һ����Ҫ�����Ŀǰ��Ȼ�����S������(zh��n)[43,44]���e������ϡҰ�����o(h��)��(d��ng)��� DNA (ancient DNA, aDNA)���о��I(l��ng)��t�������y��(2)��މ�� PCR�U(ku��)������(du��)�U(ku��)���a(ch��n)��މ�� NGS �y(c��)��ԓ��������������U(ku��)���������w����MĿ��Ƭ�Σ��ٌ��U(ku��)���a(ch��n)��ֱ���ϙC(j��)މ�� NGS �y(c��)�o(w��)�蘋(g��u)�� DNA �Ď�(k��)[45]��ԓ�����ă�(y��u)��(sh��)������Ҫ����ʼ DNA �ӱ����٣��e�m��С�����x�ͭh(hu��n)�� DNA �о��I(l��ng)���P(gu��n)�I����ģ�� DNA ���|(zh��)���� PCR ������خ��ԡ�
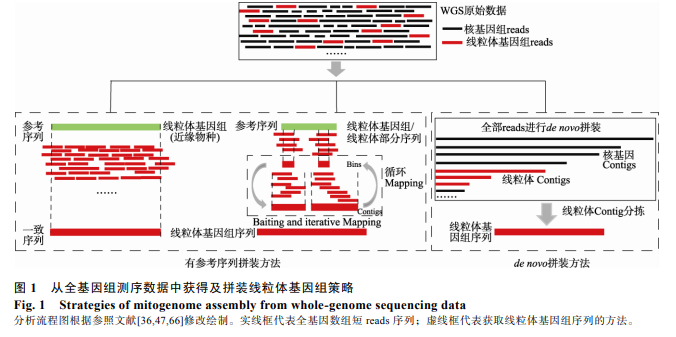
����NGS ��(sh��)��(j��)���V����(y��ng)���������ƌW(xu��)�ĺܶ��I(l��ng)����������މ������W(xu��)��Ⱥ�w�z���W(xu��)�Ƚ�ʾ��N����Դ�͔U(ku��)ɢ�vʷ���慧�]����Ҫ�����á��о��߂��������F(xi��n)�˻���(sh��)��(j��)�;����w��(sh��)��(j��)���F(xi��n)����һ�µ��Vϵ�P(gu��n)ϵ���e�Ǿ��Џ�(f��)�s��Ⱥ�w�vʷ���Ⱥ(������������z��Ư׃��ƫ�����w��������Vϵ�֒���)����Ҋ���ڷ��� NGS ��(sh��)��(j��)�r(sh��)�����˺˻���M��(sh��)��(j��)�⣬�����w����M��(sh��)��(j��)Ҳ�dz���Ҫ��Ȼ����Ŀǰͨ�^(gu��) NGS�����@�õ�ȫ����M��(sh��)��(j��)�м������˾����w����M��(sh��)��(j��)�ͺ˻���M��(sh��)��(j��)����ȫ����M��(sh��)��(j��)�У��mȻ�c�˻��� reads �Ĝy(c��)�������ȣ������w reads �Ĝy(c��)������Ǻ˻���� 100~1000 ��(��(x��)���д��ڎ�ʮ����(sh��)�ق�(g��)��ؐ) [46]�����Ǿ����w����M���� reads ��(sh��)��ֻռ�� WGS �� reads ����һ���֣����ҳ����ܵ��˻�����~�G�w(�Gɫֲ��) reads ����Ⱦ����ˣ�ʹ�ø�Ч��������Ϣ���ߺͷ������ԏĺ�����ȫ����M��(sh��)��(j��)�п��ٜ�(zh��n)�_�ث@�þ����w����M reads ��������(zh��n)�_��މ�к��m(x��)�����w����Mƴ�b���@�÷dz���Ҫ[36]�����Č����Y(ji��)��(d��ng)ǰ���õď� WGS ��(sh��)��(j��)�Ы@ȡ�����w����M���е�ƴ�b���Լ����P(gu��n)ܛ�����Ԍ�(du��)ʹ������ʹ�ò�ͬ���Ժ����P(gu��n)ܛ������o�轨�h��
����1 �Ѕ�������ƴ�b���Լ�ܛ����(y��ng)��
�����Ѕ�������ƴ�b������Ҫ�x�������N�ľ����w����M��Ƭ�����酢�����Џ��о��Ⱥ��ȫ����M��(sh��)��(j��)�в��@�����w reads������(j��)�� WGS ��(sh��)��(j��)�в��@�����w reads �Ƿ���Ҫ�����ľ����w����M���酢�����У�Ŀǰ���õIJ��Կ��Է֞飺(1)���ھ����w����(g��)����M��ƴ�b����;(2)���ھ����wƬ�ε�ƴ�b����[47,48](�D 1)���ڔ�(sh��)��(j��)���������ϣ�����ʹ��ȫ����M�Ȍ�(du��)����(�� BWA[49])���� reads ӳ�� (mapping)�������w���������ϣ�����(j��)���е������Բ��@�����w reads��Ȼ����ʹ�ò�ͬ���������L(zh��ng)���Ԍ�(du��)���@���ľ����w reads މ���������죬ֱ�����L(zh��ng)�������ľ����w����M�L(zh��ng)�ȡ�
����1.1 ���ھ����w����Mƴ�b���Լ�ܛ����(y��ng)��
�������ھ����w����M���酢�����Ы@ȡ��N��Ⱥ�w�ľ����w����M���еķ������V����(y��ng)����ϵ�y(t��ng)������Ⱥ�w�z���W(xu��)�о����� Ko ��[50]���F(xi��n)�����؈�ľ����w����M���酢�����У��@ȡ��һ��(g��) 2.2 �f(w��n)��ǰ����؈�ľ����w����M����ԭ���Ǹ���(j��)ͬԴ�Ȍ�(du��)���о��������� WGS ��(sh��)��(j��)ӳ�䵽������N�ľ����w����M�ϣ��ٸ���(j��)�����w reads �g��دB��꣬�Ķ�������е����L(zh��ng)(�D 1)���@�N�����^���@ȡ�ͅ�������Mһ�µ�����(consensus sequence)�����Ҝ�(zh��n)�_�Ըߣ��\(y��n)���ٶ��^���Ҳ���Ӌ(j��)���YԴ��
�����S���y(c��)���g(sh��)�ą�չ����(du��)��(sh��)��(j��)��������������Ҳ�����ӣ��e��������w����M�о��I(l��ng)�������މ���vʷ��������w�����ȷ�����о�[51,52]���Ƅ�(d��ng)��������w����M��ƴ�b��ע����P(gu��n)ܛ���ą�չ(�� 1)��MIA ���^������������w����Mƴ�b��ܛ�����о��ߌ�(du��)�ᰲ���ع�����^�ᵽ�� DNA މ�и�ͨ���y(c��)����ìF(xi��n)���˵ľ����w����M���酢�����У�ʹ��ԓܛ���@ȡ���ᰲ���ع���ľ����w����M[53]���S��������w����M��(sh��)��(j��)�IJ����۷e���о��I(l��ng)��IJ����U(ku��)��(du��)��(sh��)��(j��)����������ܛ���Ĺ����������Ҫ��һЩ�W(w��ng)�j(lu��)�� windows �D���Ñ������ܛ�����V��ʹ�ã����� MitoBamAnno-tator[54]��MitoSeek[55]��mtDNA-profiler[56]��mit-o-matic[57]�� MToolBox[58]��Phy-Mer[59]��mtDNA-Server[60]�� MitoSuite[61]�ȡ��@�ܛ��֧�ֶ�Nݔ���ļ���ʽ������ mtDNA-profiler �� mit-o-matic �⣬����ܛ����֧�ֶ�މ�Ƶ� Bam ��ʽ�ļ�����ˣ��@Щܛ������ֱ���xȡ��ͬܛ����ݔ����(sh��)��(j��)���ӿ�������(g��)�������̡�ֵ��ע����ǣ����Nܛ�����Ñ��x��ą�������M��(sh��)���в���� MitoBamAnnotator��mtDNA-profiler �� mit-o-matic �H�ṩ�� 1 �������M(rCRS)�� MitoSeek (rCRS, hg19)��mtDNA-Server (rCRS, RSRS) �� MToolBox (rCRS, RSRS)�ṩ�� 2 ����M��(sh��)��(j��)���� MitoSuite �ṩ�� 5 �����������M(rCRS�� RSRS��hg19��GRCh37 �� 38)��ʹ�� Phy-Mer ܛ�����Ñ������Զ��x��������M���С����⣬ͨ�^(gu��) MitoBamAnnotator��MitoSeek��MToolBox��mtDNA- Server�� mit-o-matic �� MitoSuite ܛ�����Ñ������O(sh��)������(y��ng)����(sh��)(������С��λ�����l�ʣ�MAF)��(l��i)�z�y(c��)�����w����M��׃��λ�c(di��n)�ͮ��|(zh��)��λ�c(di��n)(heteroplasmic sites, �������w����M������ͬһ��(g��)λ�ô��ڃɷN���ɷN���ϵĉA����ͣ���(l��i)Դ��������Դ��Ⱦ�������y(c��)���e(cu��)�`���خ��ԔU(ku��)����reads ƥ���e(cu��)�`�ȣ�Ҳ�����ǃ�(n��i)Դ�����w���|(zh��)�w)��MitoBamAnnotator ��Ҫ�u(p��ng)�����A(y��)�y(c��)�����w���|(zh��)��λ�c(di��n)���ڵĹ��ܣ���ʹ�ù��ܱ��^��һ��MitoSeek �� MToolBox �U(ku��)չ�˷������ܣ����������w��ؐ��(sh��)Ŀ���Ȍ�(du��)�|(zh��)�����Y(ji��)��(g��u)׃���z�y(c��)�ȹ��ܡ�MitoSeek ߀���Խ��� Circos[62]ܛ����(du��)�z�y(c��)����׃ �� މ �� �� ҕ �� �� �� �� �� �� �Y(ji��) ��(g��u) ׃ �� (structural variations, SVs)�͆κ�����׃��(single nucleotide polymorphism, SNPs)��MToolBox ��(y��u)��(sh��)���ڿ��Ԇδη�������(g��)��(g��)�w�����Ҍ�׃����Ϣӛ䛵� VCF �ļ��У������ױ�������עጡ����Ñ������\(y��n)�з�����^�� MitoSeek �� MToolBox ��һ����� Perl �����Z(y��)�Ե� Linux �\(y��n)��h(hu��n)����������Ҫ���d����(g��)��(d��)���� Perl ģ�K�ͱȌ�(du��)ܛ��(BWA)�Լ�׃���z�y(c��)ܛ��(GATK[63])����(du��)�ڷ�������Ϣ�о��������Ñ����b��ʹ���@�ܛ������(du��)�^���y��mtDNA-Server �� mit-o-matic ܛ���ǾW(w��ng)�j(lu��)�Ñ�D�η������ߣ��Ñ�����Ҫ��(f��)�s�İ��b�^(gu��)�̣��Hͨ�^(gu��)ע��(c��)���]����ς���(sh��)��(j��)��މ�з����������͔�(sh��)��(j��)��������(du��)��(ji��n)�Σ�ȱ�c(di��n)����ݔ���ļ���С�����ƣ��e�Ǹߜy(c��)����ȵĂ�(g��)�w�ς���(sh��)��(j��)�^�����������_���� MitoSuite ܛ���U(ku��)չ�˸�����(sh��)�ù��ܣ����ܸ���(qi��ng)����������w����M��ƴ�b��׃���z�y(c��)������׃��עጺ����A(y��)�y(c��)����ؐ��(sh��)Ŀ���|(zh��)���z�y(c��)���w�ȵĿ�ҕ���ȡ�MitoSuite ������������ڵ�ܛ��������Ҫ���b������(f��)�s��Ӌ(j��)��ģ�K���LjD�λ�����ϵ�y(t��ng)���ܱ����\(y��n)�е�һ�����ײ�����ܛ��������ֱ�ӏ� Bam �ļ����Ԅ�(d��ng)����һ�������к�މ��ϵ�y(t��ng)������Ⱥ�w�z���W(xu��)���о�[61]�����Ԍ�(du��)��������w����M���о��I(l��ng)���x�� MitoSuite �����Ѓ�(y��u)��(sh��)��
�����C��������ʹ���������������P(gu��n)ܛ����ȫ����M��(sh��)��(j��)�Ы@ȡ�����w����M���У����Ƚ���ȫ����M�Ȍ�(du��)ܛ�����������õ� BWA �� Bowtie/Bowtie2[64]�����Ŀ� reads �в��@�������w����M reads���@�ɷN�Ȍ�(du��)ܛ����(y��u)��(sh��)���ڿ��Ԍ�(du��) reads �e(cu��)��� reads ��̎ƥ��މ�кY�x���^(gu��)�V��ͨ�^(gu��)���m(x��)���|(zh��)�ث@ȡ�������ľ����w reads�����ǣ��o(w��)���^(q��)�� Numts �;����w��ؐ��(sh��)���Ķ�Ӱ푾����w���|(zh��)�Եęz�y(c��)�����⣬�@Щ���������P(gu��n)ܛ����Ҫ�x�������N�ľ����w����M�������У�����x��މ���P(gu��n)ϵ�^�M(j��n)����N�ľ����w����M���酢�����У���ȫ����M�Ȍ�(du��)���^(gu��)���п��ܕ�(hu��)���� reads �e(cu��)����������з����(d��o)�²��օ^(q��)��Ȍ�(du��)���϶����F(xi��n)ȱʧ��(sh��)��(j��)(gap)���Ķ�Ӱ푵����m(x��)�����w����Mƴ�b�Ĝ�(zh��n)�_�Ժ�������[38]����ˣ��x����m��N�ľ����w����M���酢��������ԓ������ܛ����(y��ng)�õ��P(gu��n)�I����(du��)��Ҫ�о�����N�o(w��)���_���������N�������Ǵ_�����������N���](m��i)�����о����w����M��(sh��)��(j��)������£��@��(g��)�������кܴ�ľ�����[36,39]��
����1.2 ���ھ����wƬ��ƴ�b���Լ�ܛ����(y��ng)��
������������������N�ľ����wȫ����M���酢�����е�ƴ�b���Լ����P(gu��n)��ܛ������(sh��)�m�����˵ľ����w����Mƴ�b��׃���z�y(c��)��׃��עጵȡ��S��Խ��(l��i)Խ��������N���о��������w����M����Ҳ���V����(y��ng)���ڷ�ģʽ��N���о���[65]���H���˵Ļ���M���酢�����е�ܛ����(l��i)�@ȡ�ͷ���������N�ľ����w����M���оͱ��F(xi��n)���ܴ�ľ����ԣ����������Ҫ�_���m�÷������V�ľ����w����Mƴ�bܛ�����c��reads ֱ��ӳ�䵽�����w����M�������е�ƴ�b������ƣ��������x���z���P(gu��n)ϵ�^�M(j��n)���^����N�ľ����w����M�����������w�������У���(l��i)މ��������N�ľ����w����M���Ы@ȡ��ƴ�b��ԓ�������Ƚ���ȫ����M�Ȍ�(du��)ܛ�����^(gu��)�V��� WGS ��(sh��)��(j��)ӳ�䵽���������ϣ��߸��w�����B�m(x��)�ľ����w reads �M�����ЉK(bins)���@Щ�Ϊ�(d��)�� bins ���߸���(j��) bins �دB����B�ӳ� Contigs ��Qԭ�ȵą������У��������´�ӳ��İ�����(baiting sequencing)�����η���(f��)�� WGS ��(sh��)��(j��)ӳ�䵽�����ɵİ����������L(zh��ng)���У�������L(zh��ng)�������ľ����w����M�L(zh��ng)��(�D 1)������(f��)ӳ�����Q�����п��Ա��Ⅲ�����к�ƴ�b������ƫ���ԡ�ƴ�b�^(gu��)������Ҫ�{(di��o)�� Kmerֵ(ƴ�b�^(gu��)���� reads�����L(zh��ng)�Ȟ� K ��һ�ι̶�����������)��С������(f��)�� WGS ��(sh��)��(j��)ӳ�䵽��������މ���������L(zh��ng)�������Ҫ���Ĵ�����Ӌ(j��)���YԴ��ԭʼ��(sh��)��(j��)Խ��Խ����Ӌ(j��)���YԴ������x���z���P(gu��n)ϵԽ�M(j��n)����N���x��İ�����Խ�̣�ƴ�b�r(sh��)���������L(zh��ng)�t��Ҫ�����ѭ�h(hu��n)�Δ�(sh��)��Ӌ(j��)��r(sh��)�gҲ��Խ�L(zh��ng)��
����Hahn ��[66]�_���� MITObim ܛ������ֱ�ӏ� WGS ��(sh��)��(j��)��ƴ�b��ģʽ��N�ľ����w����M���@��(g��)ܛ��Ƕ���� MIRA �� IMAGE Ӌ(j��)��ģ�K����� MIA�� MITObim �Ĝ�(zh��n)�_�Կ����_(d��)�� 99.5%���ϣ����؏�(f��)�^(q��)�������Ч�����a(b��) gap��Ӌ(j��)���ٶȺ̓�(n��i)������Ҳռ�Ѓ�(y��u)��(sh��)���ɞ�Ŀǰ��V��ʹ�õľ����w����Mƴ�bܛ����ԓܛ����֧���p������(paired-end reads, PE reads)��֧�� Iontorrent��454 �� PacBio �y(c��)��ƽ�_(t��i)��(sh��)��(j��)�����ҽ��hԭʼ��(sh��)��(j��) reads ��(sh��)����Ҫ���^(gu��) 20~40 ���f(w��n)�l��������������h��ԭʼ reads ���S�C(j��)��ȡ���� reads���@�Ӿ͜p�� reads �Ĕ�(sh��)�������^(gu��)�@�ӿ��ܕ�(hu��)Ӱ�ƴ�b�Y(ji��)���Ĝ�(zh��n)�_�Ժ������ԡ���(d��ng)Ȼ��MITObim Ҳ�o(w��)����Q�����w����Mƴ�b��һЩ�Ȟ��(f��)�s�Ć�(w��n)�}���� Numts����(f��)�s�ğo(w��)����(d��ng)���ֲ��ľ����wƴ�b��[67]��ARC[47]ܛ����ƴ�b�^(gu��)������� MITObim ܛ�������߶������x���H���P(gu��n)ϵ�^�M(j��n)����N�ľ����w����M���߾����w�������оͿ��Եõ������ľ����w����M���У���Ҫ�IJ�����������L(zh��ng)��ʽ�� ARC ��ֱ�ӌ�(du��) bins މ��ƴ�b������е����L(zh��ng)���� MITObim �t�Ƿ���(f��)���� reads ����������ӳ��������L(zh��ng)���С��������ȫ����Mƴ�bܛ����ARC ���nj��� reads މ�Џ��^ƴ�b��������ͨ�^(gu��)ӳ��ķ�ʽ��(du��) reads �دB�� bins މ��ƴ�b����(y��u)��(sh��)���ڲ��ă�(n��i)�棬�\(y��n)���ٶ��^�졣���⣬ARC �����ϲ��ܽ����(y��n)�ص� DNA �|(zh��)���͵��|(zh��)���� reads ��Ӱ푣��e�� aDNA�������\(y��n)���ٶȱ� MITObim �͂��y(t��ng)��ƴ�b������[47]�� Li ��[68]ʹ�� ARC ܛ����(du��) 19 ��(g��)�[�U���x(Caenorhabditis)��Nމ�о����w����Mƴ�b���y(c��)ԇ�˲�ͬ�y(c��)��ƽ�_(t��i)(Roche��454��Illumina �� Ion Torrent)��(du��)�����w����Mƴ�b��Ӱ푣��Y(ji��)�����F(xi��n) ARC ܛ����(du��) 454 ƽ�_(t��i)�Ĕ�(sh��)��(j��)މ�з����r(sh��)��(hu��)���������ܵ�ԭ���������L(zh��ng)�ȷ�����(d��o)��(sh��)��(j��)������Ҫ�^���Ӌ(j��)���YԴ������ ARC ƴ�b�������Զ�Ҫ�� MITObim �á�Ȼ���� Dierckxsens ��[47]�� ARC ܛ����(du��)��Ä�~��(Gonioctena Intermedia)މ�о����w����Mƴ�b���Y(ji��)�����F(xi��n)�M�� ARC ��(zh��n)�_�Ը�(99.99%)�������܌������wƴ�b��һ�l Contig �ϣ��������^��(���w�������w����M�� 85.39%)��
����Dierckxsens ��[38]�_���� NOVOPlasty ܛ��������� SSAKE[69]�� VCAKE[70]�㷨���������� reads ����ڹ�ϣ���У��Ա� reads �Ŀ����xȡ������\(y��n)���ٶ��^�졣NOVOPlasty ܛ����Ҫ�ṩһ�l�����У�������һ�l�� read��һ�ξ��a�������У������������ľ����w����M���С�ֵ��ע����ǣ�NOVOPlasty �c ARC ƴ�b���Բ�ͬ���ǣ�NOVOPlasty �����ṩ�İ����Џ� WGS ��(sh��)��(j��)�Ы@ȡ�����w����M��һ�l read��Ȼ���ٌ�(du��)���@���� read މ���p�����졣���ߌ� NOVOPlasty �c��(d��ng)ǰ������ƴ�bܛ������^������ MITObim��MIRA��ARC��SOAPdenvo2 �� CLCbio���Y(ji��)�����F(xi��n)������ ARC �⣬����ܛ�����������wƴ�b��һ�l Contig��ͨ�^(gu��)��(du��) NOVOPlasty ƴ�b��������މ���|(zh��)���u(p��ng)�����](m��i)�Ѕ��F(xi��n)ȱʧλ�c(di��n)�Ͳ��_���ĉA��λ�c(di��n)��������(zh��n)�_�Ժ������Ըߡ�NOVOPlasty ��Ӌ(j��)���ٶ���졢����M���w����ߣ�CLCbio ��(zh��n)�_��ͬ��Ҳ�_(d��)���� 100%�����ǻ���M�ĸ��w�Ȳ���(89.96%)�� MIRA �� ARC ���w�F(xi��n)��ߵĻ���M���w�ȣ����ǜ�(zh��n)�_����͡����Ӝy(c��)���w�Ⱥ� reads ���L(zh��ng)�ȿ������ NOVOPlasty �������Ժ͜�(zh��n)�_�ԣ��e�Ǹ��؏�(f��)�� AT �����ߵą^(q��)��NOVOPlasty �\(y��n)�в���Ҫ�d������ܛ����ģ�K����(du��)���Ñ��(l��i)�f(shu��)���b�Ͳ������^��(ji��n)��[38]��
����Ŀǰ�����~�G�w����Mƴ�bܛ��ͬ���m�Ͼ����w����M��ƴ�b������ IOGA[71]��GetOrganelle[72] �� ORG.Asm[73]�ȡ�IOGA �� GetOrganelle ����� MITObim �е�“Baiting and iterative ӳ��”�������̡� IOGA �����^(gu��)����Ҫ Bowtie2��SOAPdenovo2��SPAdes 3.0[37]�����������(l��i)���@�����w reads��ƴ�b�^(gu��)��߀��Ҫ�{(di��o)��ƴ�b����(sh��) Kmer ��С(������ 37~97)�����ͨ�^(gu��)ƴ�b��Ȼ�u(p��ng)��(assembly likelihood estimation, ALE)�ĺ��x�� Contigs ������_�������w����M[74]���@�N�����m�Ͻ���̶��^��Ę�Ʒ�ľ����w����M���~�G�w����Mƴ�b�����粩���^��Ʒ�ȡ��c����ƴ�bܛ�����^��IOGA ʹ�� ALE �z�(y��n)��(l��i)�Y�xƴ�b�õ� Contigs�����ͨ�^(gu��)�����Ȼֵ��(l��i)�Д��(y��u)��ƴ�b���С�GetOrganelle �� IOGA ��(sh��)��(j��)�������̷dz����ơ� GetOrganelle Ƕ���˪�(d��)���� Bowtie2��BLAST[75]�� SPAdes 3.0 ����ģ�K���p�� reads �͆ζ� reads (singleend reads��SE reads)���������� GetOrganelle ��ݔ���ļ���GetOrganelle ����ֱ���� SPAdes ƴ�b���^(gu��)����މ�� reads �e(cu��)�`�C�����e(cu��)���^(gu��)�V���������|(zh��)���� reads ������m(x��)�������� IOGA �� MITObim �t��Ҫ�������^(gu��)�Vܛ����ǰމ�е��|(zh��)�� reads ���^(gu��)�V�� IOGA �� GetOrganelle ƴ�bܛ����Ƕ�� SPAdes ����Ӌ(j��)��ģ�K����ƴ�b�^(gu��)������Ҫ����(f��)�{(di��o)ԇ Kmer ֵ�Ĵ�С���x����m�� Kmer ���H�܉��C�����w Scaffolds �� Contigs �������Ժ͜�(zh��n)�_�ԣ�߀���Ԝp��Ӌ(j��)��r(sh��)�g���\(y��n)�Ѓ�(n��i)��[72]��
����������S���η��Ӝy(c��)�� PacBio �� Nanopore �L(zh��ng)Ƭ�Μy(c��)���g(sh��)�ą�չ��һЩ��(f��)�s��N��ȫ����M���б��y(c��)��͑�(y��ng)�ã��e�Ƕ�w��N���؏�(f��)����N���@ʾ���L(zh��ng)Ƭ�Μy(c��)���g(sh��)�ă�(y��u)��(sh��)[27,76~80]��ͬ�r(sh��)���ѽ�(j��ng)�_������һЩ�m����ƴ�b PacBio �� Nanopore �L(zh��ng) reads ��ܛ�������� HGAP[81]��Falcon (https:// github.com/PacificBiosciences/falcon)��Canu[82]�� Sprai[83] �ȣ������@Щƽ�_(t��i)�y(c��)��õ����L(zh��ng) reads މ�о����w���~�G�w����Mƴ�b�ķ������㷨߀��ȱ����Ŀǰ�ѽ�(j��ng)��һЩ�о���ֱ��ʹ�� PacBio �� Nanopore ƽ�_(t��i)މ�о����w����M�y(c��)���މ��ƴ�b[25~29]��Soorni ��[84] ���� Perl �����Z(y��)���_���� Organelle-PBA ֱ�ӌ�(du��) PacBio ƽ�_(t��i)�y(c��)��ȫ����M�L(zh��ng)Ƭ��މ�о����w���~�G�w����M��ƴ�b��Organelle-PBA ���b��ʹ����Ҫ���b��N Perl ģ�K�Ͷ�Nܛ�������� BlasR[85]�� Samtools[86]��Blast[87]��SSPACE-LongRead[88]��Sprai �� BEDTools[89]�ȡ��mȻ PacBio �� Nanopore �y(c��)��ƽ�_(t��i)���Եõ����L(zh��ng)�� reads��������Ȼ����һ���ĉA���e(cu��)�`�ʣ������Ҫʹ�ÉA���C��ܛ��މ�ЉA���C�������� Sprai���� PacBio �� Nanopore �y(c��)��ƽ�_(t��i)����Ҫ�ڽ���(k��)���^(gu��)����މ�� DNA �S�C(j��)���͔U(ku��)�����Ҿ����x�L(zh��ng)�L(zh��ng)���c(di��n)�����Կ��������Ì������w����Mһ���Ԝy(c��)ͨ����Ч������ Numts ����Ⱦ����ͬ�r(sh��)��?y��n)?PacBio �� Nanopore �y(c��)��ƽ�_(t��i)��(du��)��Ʒ DNA �|(zh��)���ИO���(y��n)���Ҫ��Ҫ���C DNA �������ԣ����� OrganellePBA ��ʹ��Ҳ�о����ԡ�
����2 ���^(de novo)ƴ�b���Լ�ܛ����(y��ng)��
����Ŀǰ��������Խ��(l��i)Խ�����N��ȫ����M��(sh��)��(j��)�;����w����M��(sh��)��(j��)����������Ҳ�н^�����(sh��)��N�Ļ���M��Ϣ߀δ���y(c��)����ᘌ�(du��)�](m��i)�Ѕ�������M���е���N�����^ƴ�b��һ�N���ٺ͜�(zh��n)�_�ث@ȡ�z����Ϣ�IJ��ԣ��@�N�������V����(y��ng)���� DNA �� RNA ����ƴ�b�������w����M�ď��^ƴ�b�c�˻���M��ƴ�b�^(gu��)�����ƣ����ȏĺ�����ȫ����M��(sh��)��(j��)���ҵ��� reads ��һ�������У�Ȼ���ٸ���(j��)��ͬ�L(zh��ng)�ȵĴ�Ƭ���Ď�(k��)މ�� Contigs ��������B�ӣ�������L(zh��ng)�� Scaffolds ˮƽ������(j��)�����w reads �ā�(l��i)Դ��ͬ�����Է֞��ȫ����M��(sh��)��(j��)�Џ��^ƴ�b�����w����M���Ժ͏��D(zhu��n)䛽M��(sh��)��(j��)�Џ��^ƴ�b�����w����M���� (�D 1)��
����2.1 ��ȫ����M��(sh��)��(j��)�Џ��^ƴ�b�����w����M���Լ�ܛ����(y��ng)��
�������^ƴ�b�����w����M��������Ҫ�ṩ�����ľ����w����M�����w�����������酢�����С����^ƴ�b���Ȍ� WGS ��ȫ�� reads މ�Џ��^ƴ�b[47,48]�������˻���;����w���� reads ���քeƴ�b���L(zh��ng)Ƭ�����У�Ȼ������(j��)�����w����M�����L(zh��ng)�Ⱥߜy(c��)�����މ�Ї�(y��n)���Contigs�^(gu��)�V�õ����x�����wContigs�����(f��)�� WGS ��(sh��)��(j��)ӳ�䵽���x�����w Contigs �ϣ��������L(zh��ng) Contigs��ֱ�����L(zh��ng)�����������w����M�L(zh��ng)��(�D 1)���F(xi��n)�е�ܛ���� Norgal[36]�� MitoZ[39]�ȡ���(du��)��һЩ�](m��i)�н�����N�����w����M����N������ DNA �����(y��n)�صĘ�Ʒ(���� aDNA ����)�����Ѕ�������ƴ�b�������кܴ�ľ����ԡ����ԣ���(du��) aDNA ���߭h(hu��n)�� DNA ����މ�� NGS �y(c��)����މ�о����w����M���^ƴ�b����һ��(g��)�ЁY��Ч�IJ��ԡ����ǣ��@�N��������Ҫ������ȫ����M���D(zhu��n)䛽Mƴ�b��ܛ����Ӌ(j��)��ģ�K(���� SOAPdenovo2[90]��SPAdes[37]�� Velvet[91]��BIGrat[92]��CLCbio (https://www.qiagenbioinformatics.com/products/clc-assembly-cell)��SOAPdenovo-Trans[93]�� Trinity[94]��)��(du��)����(g��)����M��(sh��)��(j��)މ��ƴ�b��������Ҫ����(f��)�{(di��o)�� Kmer ֵ�ķ������_(d��)����ѵ�ƴ�b�|(zh��)�������Ժ��M(f��i)Ӌ(j��)���YԴ��Ӌ(j��)���ٶ��^����
�������y(t��ng)�ď��^ƴ�bܛ�������� SOAPdenovo2�� Newbler��SPAdes��Velvet��CLCbio��ALLPATHS[95] �� Platanus[96]�ȣ���ȫ����M����ƴ�b�^(gu��)���У��侀���w Scaffolds �� Contigs �������^(gu��)�V�������^ƴ�b�����w����M�t�������y(t��ng)�ď��^ƴ�bܛ�����ڷ����^(gu��)���п��]�����w reads �ĸߜy(c��)����ȣ������nj���h����Ŀǰ�ѽ�(j��ng)���S����(d��ng)ֲ��ľ����w����M�Ï��^��ƴ�b�����@���������ľ����w����M���С� Lee ��[97]��(du��)�۹��ƵĽ۹�(Platycodon grandiflorus) ���h��(Codonopsis lanceolata)މ���˵��w�Ȼ���M�y(c��)��Ԍ�(du��)�����w����Mމ��ƴ�b����������ʹ�� Celera��SOAPdenovo, SPAdes �� CLCbio �� 4 �Nȫ����Mƴ�bܛ����(du��)ȫ�� reads މ�Џ��^ƴ�b���õ��ɺ˻���;����w�M�ɵ� Contigs ��(k��)����θ���(j��)�����w�� Contigs �ͺ˻���M�� Contigs ƽ���y(c��)����ȵIJ�_�����x�����w Contigs���ٌ� WGS ��(sh��)��(j��)�Ȍ�(du��)�����x�����w Contigs �ϣ����ѭ�h(hu��n)��� Contig �����L(zh��ng)�����õ������ľ����w����M[97]��������@�Nƴ�b���ԣ�Al-Nakeeb ��[36]�_���� Norgal ܛ������ʹ�� MEGAHIT[98]ƴ�bܛ����(du��) NGS ��(sh��)��(j��)މ�Џ��^ƴ�b��Ȼ���ٌ� NGS ��(sh��)��(j��)����ӳ�䵽ƴ�b�õ� Contig �ϣ�ͨ�^(gu��)�����w�ͺ˻���M�� reads ���w�ȁ�(l��i)�Дྀ���w Contig(s)������ͨ�^(gu��)�c������ͬ���Եľ����w����Mƴ�bܛ�����^���F(xi��n)��Norgal ܛ���Ĝ�(zh��n)�_�Ժ� NOVOPlasty ܛ�����ƣ����Ǐ��\(y��n)���ٶ��ρ�(l��i)���^�� NOVOPlasty �M(j��n)�� Norgal �� MITObim Ҫ�죬ԭ���� Norgal ��Ҫ�{(di��o)����ͬ Kmer ��С��(du��)����(g��)����Mމ��ƴ�b��Ȼ���ٱȌ�(du��) reads ��Ӌ(j��)��˻���M reads �Ĝy(c��)����ȁ�(l��i)�Д�ƴ�b�Ŀɿ���[36]��
�������P(gu��n)֪�R(sh��)���]�������w����MՓ����ʲô�о��ɹ�
�����S���Ñ�(du��)��(sh��)��(j��)����������Խ��(l��i)Խ��Ҫ��(ji��n)������Ч�ʵĔ�(sh��)��(j��)�������̡�����ȫ������õ��Ñ��w�(y��n)��ܛ��Խ��(l��i)Խ�ɞ����е���Ҫ��Meng ��[39] �_���� MitoZ ܛ������“һ�Iʽ”�،�(du��)�����w����Mމ��ƴ�b��עጺͿ�ҕ����ԓܛ�������˶�NӋ(j��)��ģ�K������ԭʼ��(sh��)��(j��)���A(y��)̎�������^ƴ�b�����x�����w���еĸ����;����w����M��עጺͿ�ҕ���ȹ��ܡ����������ܛ����ԓܛ���܌�(du��)���|(zh��)���� reads���A������ȱʧ�� reads �ͽ���(k��)�� PCR ����� reads މ���^(gu��)�V���Ա��C���m(x��)������(sh��)��(j��)�Ŀɿ��ԡ�MitoZ ������ SOAPdenovo-Trans ���㷨���ĺ˻���M�е� reads މ�о����w����M�ď��^ƴ�b����ԭ���ǣ�����(j��)�����w����M reads ��ƽ���y(c��)������M(j��n)�Ⱥ˻���M�ĸߣ��O(sh��)�ò�ͬ�� Kmer ����(sh��)��(l��i)�_(d��)����ѵ�ƴ�bЧ�����@��(g��)ܛ���ṩ�˃ɷNƴ�b��ʽ�����ģʽ(quick model)�Ͷ� Kmer ģʽ������(j��)���ߵĽ��h�M����ʹ�ö� Kmer ģʽ�{(di��o)����ͬ Kmer ����(sh��)���Ա��C��(f��)�s�����w����Mƴ�b�������Ժ͜�(zh��n)�_�ԡ���ƴ�b�Ļ���(sh��)�������еĿ��L(zh��ng)�ȷ���މ�б��^��MitoZ ���Ѕ������е�ƴ�b���Ը����Ѓ�(y��u)��(sh��)���e�nj�(du��)����N�g���ƶȺܵ͵Ļ����⣬���˸��ܛ���㷨�IJ���؏�(f��)���С�AT �����ͮ��|(zh��)����(���|(zh��)��λ�c(di��n)ռ��׃��λ�c(di��n)�Ĕ�(sh��)��)��Ҳ��Ӱ푾����w����M��ƴ�b�����Ժ͜�(zh��n)�_�Ե��P(gu��n)�I����[39]��MitoZ ��(du��)�����w����M��ע�(Blast��Genewise��MiTFi �� Infernal)�Լ���ҕ��(Circos)���ܼ��������������ܛ��ģ�K������g�ӵ�?c��i)U(ku��)չ��ƴ�bܛ���Ĺ��ܣ�Ҳ�O��غ�(ji��n)���˔�(sh��)��(j��)�ķ����^(gu��)�̡�
����2.2 ���D(zhu��n)䛽M��(sh��)��(j��)�Џ��^ƴ�b�����w����M���Լ�ܛ����(y��ng)��
������һ���y(c��)���g(sh��)�ą�չͬ�r(sh��)�Ƅ�(d��ng)���D(zhu��n)䛽Mˮƽ���о������D(zhu��n)䛽M��(sh��)��(j��)�Ы@�û���M���a�����ѽ�(j��ng)�ܳ��죬������ RNA �D(zhu��n)䛱��а��������ľ����w���a�����D(zhu��n)䛱��������о����_���˿��Ը�Ч�؏��D(zhu��n)䛽M��(sh��)��(j��)�и��������w���a�������е�һЩܛ�����@Щ������ԭ���Ǹ���(j��)�����w�ڼ�(x��)����(n��i)�ؐ��(sh��)�������������w���a���� mRNA �� reads �y(c��)������M(j��n)�Ⱥ˻���M�ľ��a���� reads �ߣ����и�ˮƽ�Ļ�����_(d��)����Plese ��[99]�_���� Trimitomics ܛ���ܿ�����Ч�Ï��D(zhu��n)䛱� reads ���挦(du��)�����w���a��������މ��ƴ�b��ԓܛ���ķ������̰����� NOVOPlasty�� Bowtie2/Trinity �� Velvet �� 3 ��(g��)��(d��)��ƴ�b�^(gu��)�̣�(1)����ʹ�� NOVOPlastyܛ����ȫ���� RNA readsމ�Џ��^ƴ�b������(j��) Kmer ��С����(25��39��45 �� 51)�_�������w���a���е�������;(2)����](m��i)��ƴ�b�������ľ����w���a���л���ƴ�b���������У��t��ʹ�� Trimmomatic 0.33[100]��(du��)ԭʼ RNA readsމ���^(gu��)�V������ Bowtie2[64]ܛ�����^(gu��)�V��� reads �Ȍ�(du��)��������N�ľ����w����M�ϣ��� Trinity[94,101]��(du��) mappedread މ�Џ��^ƴ�b;(3)ʹ�� Velvet ܛ����(du��)ȫ�����D(zhu��n)䛱�މ�Џ��^ƴ�b�������� BlastN ܛ��[102]�_���õ��ľ����w Contigs��������� 3 �N�������](m��i)��ƴ�b�������ľ����w���a���У���ô��ʹ�� Geneious ܛ���������� 3 �N����ƴ�b�ĽY(ji��)�����ٌ����ϵĽY(ji��)���� NCBI ��(sh��)��(j��)��(k��)��މ��ͬԴ���b��������ͨ�^(gu��)��(du��) 6 ��(g��)�o(w��)����(d��ng)��މ�о����w���a�����ƴ�b���Y(ji��)�����F(xi��n) 3 �Nƴ�b�^(gu��)�̶��܉��w�� 97%���ϵľ����w���a�������С���ƴ�b�����Ժ͜�(zh��n)�_�ԁ�(l��i)�u(p��ng)�� NOVOPlasty�� Bowtie2/Trinity �� Velvet ƴ�b�^(gu��)�̵Ŀɿ��ԣ��Y(ji��)�����F(xi��n) 3 �Nƴ�b��������N�������� A.valida �� P.dumerilii �@�ɷN�~�΄�(d��ng)�Bowtie2/Trinity ƴ�b���̵õ��ľ����w���a���е��|(zh��)�����á������\(y��n)�Еr(sh��)�g���\(y��n)�Ѓ�(n��i)���ϱ��^��NOVOPlasty ƴ�b���̸����Ѓ�(y��u)��(sh��)��ֵ��ע����ǣ�Trimitomics ܛ���ṩ 3 �Nƴ�b���̣�ͨ�^(gu��)�Д�ƴ�b�Y(ji��)���������ԁ�(l��i)�Д��Ƿ�މ������ƴ�b���̡�ͬ�r(sh��)��(du��)�ڏ�(f��)�s��N�ľ����w����M��߀�������� 3 �Nƴ�b���̵ĽY(ji��)���������˿ɿ��ԡ� ——Փ�����ߣ����l(w��i)������