�l(f��)���r(sh��)�g��2022-05-23���ٷ��늹��QՓ���g�[��1��
ժ Ҫ�� ժҪ��ճ�Y(ji��)�������늳�늘O�������Ҫ���ϣ����������m�٣��s�ǘO�����W(xu��)���ܵ���Ҫ�Г�(d��n)�ߡ��F(xi��n)�еď�(f��)��늘O���W(xu��)�О�ģ�Ͷ���ճ�Y(ji��)���Ӽ��O(sh��)�龀���Բ��ϣ��@���y����ճ�Y(ji��)���ӵď�(f��)�s���W(xu��)�О飬��˞�����������O��{��(j��)�w���cճ�τ��ڳ����^�̵����W(xu��)�О飬����(j��)���е�
����ժҪ��ճ�Y(ji��)�������늳�늘O�������Ҫ���ϣ����������m�٣��s�ǘO�����W(xu��)���ܵ���Ҫ�Г�(d��n)�ߡ��F(xi��n)�еď�(f��)��늘O���W(xu��)�О�ģ�Ͷ���ճ�Y(ji��)���Ӽ��O(sh��)�龀���Բ��ϣ��@���y����ճ�Y(ji��)���ӵď�(f��)�s���W(xu��)�О飬��˞�����������O��{��(j��)�w���cճ�τ��ڳ����^�̵����W(xu��)�О飬����(j��)���еČ�(sh��)�(y��n)��(sh��)��(j��)�M�ϵõ��M�Ͼ����^�ߵ�ճ���Ա���(g��u)ģ�ͣ������c��x�ӔU(ku��)ɢ������ʽ���ƵĂ��᷽������ģ�M���Թ��w���ڳ�늷���^�̵đ�(y��ng)��׃�����о��Y(ji��)���������w��䇻��r(sh��)��Ҫ������׃�ΰl(f��)���ڵ�һ��(g��)��늷��ѭ�h(hu��n)�^���У�ճ�Y(ji��)�ӵ����(y��ng)��׃����Ƽ{���w���g�đ�(y��ng)��׃�����S��ճ�Y(ji��)����ȵ����ӣ�ճ�Y(ji��)�ӽ�������(y��ng)����(hu��)��u����;߀�У������(y��ng)׃��ճ�����Ա���(g��u)ģ�͌�(du��)��ճ�Y(ji��)��PVDF��׃�О��������ھ����Ժ� NeoHookean������ģ�͡�
�����P(gu��n)�I�~��ճ�Y(ji��)��;�{���w��;ճ���Ա���(g��u)ģ��;��늷��ѭ�h(hu��n);��(y��ng)��
������x��늳�����ɳ��늳��ڄ�(d��ng)��늳��I(l��ng)��l(f��)չѸ�٣�ʹ������������ѭ�h(hu��n)�Δ�(sh��)늳ص�����Ͱl(f��)չ���l(f��)ͻ������x��늳غ��ĽM�ɲ�������ؓ(f��)늘O���Բ��Ϻ�늽�Һ��늳س�늹����^������x�ӏ����O����Ó��Ƕ��ؓ(f��)�O���ϵ��^�̣�������^���c֮�෴�����g��x�Ӳ��ƭh(hu��n)늘O���Բ��ϵľ���Y(ji��)��(g��u)���H�����g���׃������(d��o)�»��Բ��Ϻ��^�w�e��׃�����Ķ���늳�(n��i)���a(ch��n)����(y��ng)����
����ճ�Y(ji��)����늳���ؓ(f��)�O�������^�̵���Ҫ���ϣ��c�������ϰ�һ���������w����늘O���Բ���ճ�Y(ji��)�����γ�늘O��(f��)��Ĥ��ʹ늘O���Բ��ϡ���(d��o)늄���늘O�����w֮�g�������õ���ӌ�(d��o)늾W(w��ng)�j(lu��)�����о��@ʾճ�Y(ji��)����(du��)��x��늳ص�ѭ�h(hu��n)��������Ҫ��Ӱ�[1]�����S���о��۽����M(j��n)�F(xi��n)�е�ճ�Y(ji��)�������_�l(f��)�µ�ճ�Y(ji��)���䷽��Munaoka��[2]���������Ͼۺ�������ճ�Y(ji��)�����ڸ�����x��늳ع�ؓ(f��)�Oѭ�h(hu��n)����;Rajeev��[3]�_�l(f��)�˚����ǽ�֦�۱���������(CS-g-PANIs)�����ꖘO�ۺ���ճ�τ�����;Yang��[4]���˱��ָ���ӂ���(d��o)�ԡ��C(j��)еճ���ԡ���չ�Ժ�늽��|(zh��)�����_�l(f��)��һ�N���ж�ܵ�ճ�Y(ji��)���ۺ�����@Щ�о������P(gu��n)עճ�ӄ��Ļ�����(y��ng)����(y��ng)׃�О顣ճ�Y(ji��)������x��늳��е������mȻ�^�٣������s������(g��)늘O���W(xu��)���ܵ���Ҫ�Г�(d��n)�ߣ���(du��)늘O�����a(ch��n)�����늳ص�늻��W(xu��)��������ҪӰ�[5-6]��ճ�Y(ji��)���ęC(j��)еʧЧ�������x��늳�늘O����˥�p����Ҫԭ��֮һ���ڳ��/����^���У������w�����w�e׃����(hu��)��ճ�Y(ji��)��ճ�Y(ji��)��̎�����@���đ�(y��ng)����(y��ng)׃[7]���_(d��)��ijһ�ֵ�r(sh��)��ճ�Y(ji��)������(j��ng)�v����׃�Σ���(d��o)��һЩ�������Ӯa(ch��n)��늸��x����ֹ�����e�O���c늻��W(xu��)����(y��ng)���Ķ���(d��o)�²������늳������pʧ����(d��ng)ǰ�Ķ�Nճ�Y(ji��)���У���ƫ����ϩ(PVDF)���^�õĿ�����߀ԭ�������᷀(w��n)���Ժ���ɢ�ȃ�(y��u)�c(di��n)����Ŀǰ��x��늳�����õ�ճ�Y(ji��)����
����늘O���ϣ���Ȃ��y(t��ng)��ʯī��늘O(��������372mAh/g)������и��ߵ���Փ������3579mAh/g��������ă�(y��u)�|(zh��)���ܶȃ�(ch��)��ؓ(f��)�O���ϡ�Ȼ�������Ƕ��/Ó������(y��ng)�У���Ĵ��w�e��Û(�s300%)�տs���Բ��H��(hu��)��(d��o)��(f��)��늘O�����w����(n��i)�a(ch��n)����(y��ng)�����������܌�(d��o)�»����w�����ѣ�ճ�Y(ji��)���Ľ��愃�x����K��(d��o)��늳�ʧЧ��һֱ���Ƽs��V��ʹ�õ���Ҫ�ϵK��
�������P(gu��n)֪�R(sh��)���]��늳ع̑B(t��i)늽��|(zh��)Փ�İl(f��)���ڿ����]
����Ŀǰ�_չ��ճ�Y(ji��)��������о���Ҫ�nj�(sh��)�(y��n)�y(c��)ԇճ�ӏ�(qi��ng)�ȣ���˺�ѺͿ̄��y(c��)ԇ[7-9]���@�N���^�߶ȵĜy(c��)ԇ�������붨�����u(p��ng)�r(ji��)ճ�Y(ji��)��(qi��ng)��[10-12]�����nj�(du��)�����{�׳߶ȵĻ����w���g��ճ�Y(ji��)��(qi��ng)�Ⱥ��yӋ(j��)�㡣�о��l(f��)�F(xi��n)�{���w��ֱ��С��150nm �r(sh��)��늻��W(xu��)ѭ�h(hu��n)�^�������m��(y��ng)��Ĵ��w�e��Û���װl(f��)���w������[13]��Hertzberg���ˈ�(b��o)������x��Ƕ��/Ó����һ��(g��)�����й���ڼ{���g��(n��i)�Π�IJ�����׃��[14]��Takahashi��[15]��ճ�Y(ji��)���Ӽ��O(sh��)�龀���Բ��ϣ��о���ʯī/��ƫ����ϩ��(f��)��늘O�ęC(j��)еʧЧ��Higa[16]�о���Ƕ䇹��w���cճ�Y(ji��)����֮�g�����W(xu��)����ã�Lee��[17]�Y(ji��)��늻��W(xu��) ���W(xu��)ģ�ͺ̓�(n��i)�ۅ^(q��)ģ���о�Ƕ��^���еĽ���Óճ��Takahashi��[18]�о���ʯī/��ƫ������ϩ(PVDF)��(f��)��늘O�ęC(j��)еʧЧģʽ;Lee��[19]�о���ճ�Y(ji��)��������(du��)늘O�C(j��)е푑�(y��ng)��Ӱ푡������о�����ճ�Y(ji��)���Ӽ��O(sh��)��龀���Բ��ϣ��@�Ӽ��O(sh��)��(hu��)ʹӋ(j��)��Y(ji��)�����F(xi��n)һ��ƫ�����?x��)�����?j��)��Y(ji��)���c��(sh��)�H�������Mattos[20]�Č�(sh��)�(y��n)����늳�����õ�ճ�Y(ji��)�� PVDF���Ǻ��εؾ������О飬�����Џ�(qi��ng)�ҵķǾ������c(di��n)��Nadimpalli[21]�Č�(sh��)�(y��n)�y(c��)������ PVDF���Ϗ�ճ����ģ�ͣ���ģ�M�о�������ճ�Y(ji��)��̎��龀����ģ�͕�(hu��)����(d��o)��(f��)��늘Oģ�͵�Ӌ(j��)�㲻��(zh��n)�_��Ҳ���о����� Neo-Hooke������ģ������ճ�ӄ������W(xu��)�О�[22-23]��Vinogradov��[24]��(j��ng)�^�y(c��)ԇ�l(f��)�F(xi��n) PVDFճ�Y(ji��)������ճ�����О飬���nj������^�̰l(f��)������׃��ճ�Y(ji��)�������(f��)��늘O��Ӌ(j��)��ѭ�h(hu��n)���ܵ��о��^�١�����x����w���c PVDFճ�Y(ji��)�������о���(du��)��ͨ�^����Ԫ�������о���x��늳��ڳ��/����^���л��Բ��Ϲ��ճ�Y(ji��)�� PVDF�đ�(y��ng)����(y��ng)׃�ݻ����Ա��ҵ��m������ճ�Y(ji��)����׃�ı���(g��u)ģ�ͣ�̽��ճ�Y(ji��)��������(du��)��ճ�Y(ji��)��/����摪(y��ng)����Ӱ푣��M(j��n)һ��������x��늳ع���ʧЧ�C(j��)���������ʹ�É�����ָ��(d��o)ճ�Y(ji��)���x�����������ճ�Y(ji��)�����ϵ��_�l(f��)��
�����mȻ�����Լ��O(sh��)Ӌ(j��)�㷽�㣬����(du��)���^��(f��)�s��ճ�Y(ji��)�����e�Ǵ��w�e��Û�տsؓ(f��)�O�裬���g��ճ�Y(ji��)�������s׃�����^���ܳ��^80%���ٲ����m����С��׃�ľ����Լ��O(sh��)�@���^�ں��Σ������Է���(y��ng)ճ�Y(ji��)������׃�W(xu��)�О飬��������`��Ķ���(d��o)�½Y(ji��)������(zh��n)�_�ԣ�����о��x��ճ���ͳ����Ա���(g��u)���̣�ͬ�r(sh��)�c�����Ԍ�(du��)�ȣ�����(j��) PVDF�����W(xu��)��(sh��)�(y��n)[26]���䑪(y��ng)����(y��ng)׃�����M�ϣ��õ����� PVDF�ľ����Ա���(g��u)ģ�ͅ���(sh��)����ģ���Ͳ��ɱȡ�Neo-Hookean�����Ա���(g��u)ģ�ͅ���(sh��)��ճ���Ԕ�(sh��)ģ�ͅ���(sh��)�����1��ʾ���ı��п��Կ�������prony��(j��)��(sh��)��ʾ��ճ����ģ����3��(g��)ģ�����c��(sh��)�(y��n)�������Ǻ���ã�����(du��)�`����С������ԓģ�����ό�(sh��)�H��
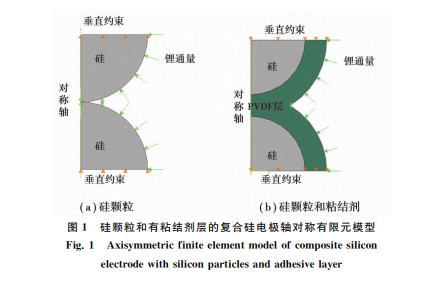
����3 ���w����ճ�Y(ji��)���ڳ�늺ͷ���^�̵�����Ԫģ�M
��������Ӌ(j��)���(f��)�Ϲ�늘O�w����ճ�Y(ji��)���ڳ���ѭ�h(hu��n)�^���еđ�(y��ng)���������]���w������Û���տs�a(ch��n)���đ�(y��ng)��������䇵IJ�����U(ku��)ɢ���ٶ�ճ�Y(ji��)���Ӿ���ķֲ���2��(g��)�����w��֮�g;Ӱ�䇔U(ku��)ɢ������ֻ����x�ӝ�ȣ���䇝���ݶȵ���������x�����ɔU(ku��)ɢ��ȡ�U(ku��)ɢ����(sh��)[26]D=10-16(m2·s-1);���ڌ�(sh��)�H�е�ճ�Y(ji��)�������������T����Ȳ�ڵČ�(d��o)����ӄ������ұ�늽�Һ���ݣ�����J(r��n)��ճ�Y(ji��)�������õ���x�ӌ�(d��o)�w������x��ͨ�^ճ�Y(ji��)������ȫ�]����K�����w���������x�ӝ���cճ�Y(ji��)�����ӵ���x�ӝ����ͬ��������D1��ʾ���S��(du��)�Q����Ԫģ�͡��xȡ�����w���돽[31]�� R=100nm��ճ�Y(ji��)���Ӻ�40nm��Ӌ(j��)��㶨ͨ����x�ӔU(ku��)ɢ��r��
����3.1 ���w���ڳ�늷���^�̵đ�(y��ng)��׃��
�����ڹ��w������ʩ�Ӻ㶨���ͨ���������Ҝ��µ���Փ����Cmax=29.89×109 C/m3����0.0965A/m2 ������ܶȌ���䇻���ƽ�����C=0.425������^���й��w���� Mises��(y��ng)��׃����D2��Mises��(y��ng)���ǻ���3��(g��)����(y��ng)���ĵ�Ч��(y��ng)�������w�����ɶ˵İ돽R1���w���g���|�돽R2 ��3�γ�늷��ѭ�h(hu��n)�е�׃��Ҋ�D3�͈D4��ʾ�����w���g�Ľ��|��(y��ng)��Ӌ(j��)��Y(ji��)��Ҋ�D5��ʾ�����|��(y��ng)������?y��n)��w�����|��ķ�������
������(d��ng)2�w����D����(y��ng)�����^������(y��ng)���r(sh��)���ɻ��Թ��w�����(hu��)�l(f��)�����õ�����׃����D2(c)��(d)��(e)�����ǰ���ɶ˰돽R1�����|�돽R2 �c���ӵij�ʼ�돽R ��ȣ��S������^���M(j��n)�й��w�������ɶ˰돽R1��Û�������|�돽�����w�����|����Ĵ��ڻ������ֲ�׃��Ҋ�D3�͈D4��ʾ�����ѭ�h(hu��n)�е�2�w���g���Ľ��|��(y��ng)���l(f��)���ڵ�һ��(g��)ѭ�h(hu��n)�У��S��ѭ�h(hu��n)�е������|��(y��ng)���^��һ��ѭ�h(hu��n)С��Ҋ�D5��ʾ���S��䇝��������w����Û׃����(d��o)���w���g����õĽ��|��(y��ng)��׃�����ڵ�һ�γ���^����䇝������ʹ��Û�w����D���ӄ������|��(y��ng)�����F(xi��n)�鼱�������_(d��)����ֵ�����|��(y��ng)�����^������(y��ng)�����(hu��)�l(f��)�����w����������׃�Σ��a(ch��n)�������p��;�S������^��䇝�ȵ��½������|��(y��ng)�����ͣ���(d��ng)��D���Ĺ��w��Ó�x���|�r(sh��)�����|��(y��ng)��Ѹ���½�������늳صĵ�һ�γ���^������Σ�U(xi��n)���^�̣��ڵ�һ�γ���^�̵Ĺ��w��ģ��������D���a(ch��n)���ĺܴ���|��(y��ng)����(hu��)��������(g��)늘O��������(g��)�늳ض���(hu��)�a(ch��n)����(y��ng)�����@�O�����l(f��)�����w���ķ��������ѣ�����늳ص�ʧЧ��������ԑ�(y��ng)׃���H��(hu��)ʹ늳�������ɓpʧ��߀���ܕ�(hu��)���늳�(n��i)������֦��������һϵ�І��}��
����3.2 ճ�Y(ji��)��/�w������đ�(y��ng)��׃��
������늷���^����ճ�Y(ji��)����3�N��׃����(g��u)ģ��(�����ԡ������Ժ�ճ����)��Ӌ(j��)��õ��đ�(y��ng)���͑�(y��ng)׃�����w���cճ�Y(ji��)�����|����ĺ�ȽY(ji��)���քeҊ��D6���D9��ʾ���@��քe��2.79×10-2A/m2��8.37×10-2A/m22�N����ܶȌ���䇻���ƽ���wһ�����C=0.083��C=0.238��ճ�Y(ji��)���ӵĺ��ָ2��(g��)���w���g������ľ��x��ճ�Y(ji��)�ӳ�ʼ���h0 ��40nm��
�����ĈD6ճ�Y(ji��)�ӽ��|��(y��ng)���ĽY(ji��)�����Կ�����3�N��׃ģ���о�����ģ�͵đ�(y��ng)��׃���ܴ�Neo-Hooke������ģ�͵õ��đ�(y��ng)��׃����֮��ճ����ģ�͵õ��đ�(y��ng)��׃����С���ĈD6(a)���Կ�������^��t=5s�r(sh��)�������Ա���(g��u)����Ӌ(j��)��õ��� Mises��(y��ng)���s�dz����Ա���(g��u)���̽Y(ji��)����2������Ҋ�x�ò�ͬ�ı���(g��u)���̌�(du��)��(y��ng)���A(y��)�y(c��)��(hu��)�a(ch��n)���ܴ��Ӱ푣����÷���ճ�τ���׃�О�ı���(g��u)���̺���Ҫ���D6��ճ����ģ�͵õ��đ�(y��ng)��׃��С�����С����(j��)�c��(sh��)�(y��n)�Y(ji��)�����[23]������x��ճ����ģ������ PVDFճ�Y(ji��)������׃�О���^���ό�(sh��)�H������׃�^��r(sh��)����ճ���Ա���(g��u)���̱Ȳ����m��С��׃�ľ����Ա���(g��u)���̸�������ճ��ģ��׃�ճ�Y(ji��)�����ܵđ�(y��ng)��С�ڳ�����;������׃�О��ճ�Y(ji��)�����@һ�c(di��n)�ɹ��늳�ճ�Y(ji��)���x��r(sh��)������
�����ĈD7���D8���Կ������ƽ��䇻��̶�C=0.083�r(sh��)�����ʹճ�Y(ji��)���ӽ�(j��ng)�v�^���׃�Σ�ճ�Y(ji��)�ӵĉ��s����80%���Ķ�ʹճ�Y(ji��)���ӳ��F(xi��n)���^��đ�(y��ng)���������ڿ����ײ����|�c(di��n)�����ĈA�h(hu��n)�΅^(q��)��a(ch��n)���đ�(y��ng)���͑�(y��ng)׃���ڵײ����|�����c(di��n)�đ�(y��ng)���͑�(y��ng)׃��Ҋ�D8��9(c)��(h)t=10s��Ӌ(j��)��Y(ji��)�����Խ��ճ�Y(ji��)���ȏ����h(hu��n)�(y��ng)׃λ���_ʼÓ���ԭ�����ճ�Y(ji��)���ď���С���͕�(hu��)��ճ�Y(ji��)��/�����̎�a(ch��n)���ܴ�Ľ��|��(y��ng)����Ӱ�ճ�Y(ji��)�����w���g����C(j��)е�p�����ܣ���Ҋճ�Y(ji��)�������܌�(du��)���摪(y��ng)�����@��Ӱ푣��o�����W(xu��)���܃�(y��u)����ճ�Y(ji��)�����늳ؼ��g(sh��)�M(j��n)������ġ�
�����ĈD9���D6�ĽY(ji��)�����Կ������������ʌ�(du��)��(y��ng)������(y��ng)׃��С��Ӱ푡������l����ͬ�r(sh��)���^���������µ�ճ�Y(ji��)����(y��ng)���͑�(y��ng)׃Ҫ���ڳ���������ĽY(ji��)����
����ճ�Y(ji��)��������늳����a(ch��n)��1��(g��)��Ҫ��������(y��u)��ճ�Y(ji��)��������(hu��)���L늳ص�ʹ�����ޡ���2��ʾ��������ܶ�2.79×10-2A/m2 �²�ͬPVDFճ�Y(ji��)���Ӻ��40nm��50nm��60nm��70nm��80nm �r(sh��)������ͬճ�Y(ji��)�������r(sh��)ճ�Y(ji��)�������|��(y��ng)�������Ľ��|��(y��ng)����Ȼ�l(f��)���ڳ���ѭ�h(hu��n)�ĵ�һ�γ���^���С����Կ������S��ճ�Y(ji��)���Ӻ�ȵ����ӣ�ճ�Y(ji��)�ӽ�������(y��ng)����u���ͣ�����(y��ng)�����ͷ��s��u�pС������ճ�Y(ji��)����ǻ��Բ��ϣ���(hu��)����늳صĿ�?c��)����������ڲ�����늳��������ӿ��(c��)����p�µ�ǰ���£����m��(d��ng)����ճ�Y(ji��)����ȁ���������(y��ng)�đ�(y��ng)��������C(j��)еʧЧ��
����4 �Y(ji��) Փ
����ᘌ�(du��)�늳؏�(f��)��ؓ(f��)�O�OƬ��2��(g��)���ι��w���Լ����� PDVF��ճ�Y(ji��)���ăɷN���Σ���������Ԫ�ض� λ�����ģʽ��ģ�M��䇻����w�������c PVDFճ�ӌӽ�����ڳ�늷��ѭ�h(hu��n)�^���еđ�(y��ng)����׃�Σ��õ��ĽY(ji��)Փ���£�
�������w��䇻��r(sh��)��Ҫ������׃�ΰl(f��)���ڵ�1��(g��)��늷��ѭ�h(hu��n)�^���У��@Щ��׃�������w���Π�IJ�����׃�����w���������Π�׃����(hu��)Ӱ��S��ѭ�h(hu��n)�еđ�(y��ng)���ݻ������w���w�e��Û�^���(hu��)ʹճ�Y(ji��)���Ӯa(ch��n)���^���׃�Σ�������ճ�Y(ji��)���әC(j��)е�����½�;���늕r(sh��)2��(g��)���w���Ľ��|����ֵҲ�l(f��)���ڵ�1�ε�䇻�/Ó�ѭ�h(hu��n)�У��S���ѭ�h(hu��n)�������|��(y��ng)����(hu��)�½��������1�ε�ѭ�h(hu��n)��(y��ng)�����^������(y��ng)�����п��܌�(d��o)���w����������늳�ʧЧ��ճ�ӌ��c�w���g��������(y��ng)��׃���c�����w���g���|��(y��ng)��׃����ƣ����(y��ng)����Ȼ�l(f��)���ڳ���ѭ�h(hu��n)�ĵ�1�γ���^���У��S��ճ�Y(ji��)������ȵ����ӣ�ճ�Y(ji��)�ӽ�������(y��ng)����u���ͣ���1��֮���ѭ�h(hu��n)�Б�(y��ng)�����ͷ��ȕ�(hu��)��u�pС����˿����ڲ�����늳��������ӿ�?c��)����p�µ�ǰ���£��m��(d��ng)����ճ�ӄ��ĺ�ȁ���������(y��ng)�đ�(y��ng)���������늳ؙC(j��)еʧЧ����(du��)�� PVDF��ճ�Y(ji��)������ճ����ģ�������䌍(sh��)�H����׃�О飬�@���^����׃���Ȟ���ˡ�——Փ�����ߣ�������1�������2���� ��(qi��ng)1�������1���� �t1���ں��
���������īI(xi��n)��
����[1]MagasinskiA��ZdyrkoB��KovalenkoI��etal.TowardefficientbindersforLi-ionbatterySi-basedanodes��polyacrylicacid[J].ACSAppliedMaterials&Interfaces��2010��2(11)��3004-3010.
����[2]MunaokaT��YanXZ��LopezJ��etal.Ionicallyconductiveself-healingbinderforlowcostSimicroparticlesanodesinLi-ionbatteries[J].AdvancedEnergyMaterials��2018��8(14)��1703138.
����[3]RajeevK K��KimE��NamJ��etal.Chitosan-grafted-polyanilinecopolymerasanelectricallyconductiveandmechanicallystablebinderforhigh-performanceSianodesinLi-ionbatteries[J].ElectrochimicaActa��2020��333��135532.
����[4]Wu M Y��XiaoX C��VukmirovicN��etal.Towardanidealpolymerbinderdesignforhigh-capacitybatteryanodes[J].JournaloftheAmericanChemicalSociety��2013��135(32)��12048-12056.
����[5]����╣����壬�R�ڣ���.��x��늳ع��ؓ(f��)�Oճ�Y(ji��)���l(f��)չ�F(xi��n)��[J].���W(xu��)�W(xu��)��(b��o)��2019��77(1)��24-40.WangX Y��Zhang Y��MaL��etal.Recentdevelopmentonbindersforsilicon-basedanodesinlithium-ionbatteries[J].ActaChimicaSinica��2019��77(1)��24-40.(inChinese)
����[6]LeeSH��LeeJH��Nam D H��etal.Epoxidizednaturalrubber/chitosannetworkbinderforsiliconanodeinlithium-ionbattery[J].ACSAppliedMaterials&Interfaces��2018��10(19)��16449-16457.
����[7]LeeJH��PaikU��HackleyV A��etal.Effectofpoly(acrylicacid)onadhesionstrengthandelectrochemicalperformanceofnaturalgraphitenegativeelectrodeforlithium-ionbatteries[J].JournalofPowerSources��2006��161(1)��612-616.
����[8]ParkH K��KongBS��OhES.Effectofhighadhesivepolyvinylalcoholbinderontheanodesoflithiumionbatteries[J].ElectrochemistryCommunications��2011��13(10)��1051-1053.
����[9]YooM��F(xi��n)rankC W��MoriS��etal.Effectofpoly(vinylidenefluoride)bindercrystallinityandgraphitestructureonthemechanicalstrengthofthecompositeanodeinalithiumionbattery[J].Polymer��2003��44(15)��4197-4204.
����[10]BurnettPJ��RickerbyDS.Therelationshipbetweenhardnessandscratchadhession[J].ThinSolidFilms��1987��154(1/2)��403-416.
����[11]DavanlooF��CollinsCB��KoivusaariKJ.Scratchadhesiontestingofnanophasediamondcoatingsonsteelandcarbidesubstrates[J].JournalofMaterialsResearch��1999��14(8)��3474-3482.
����[12]FuRJ��XiaoM��ChoeSY.Modeling��validationandanalysisofmechanicalstressgenerationanddimensionchangesofapouchtypehighpowerLi-ionbattery[J].JournalofPowerSources��2013��224��211-224.
����[13]LiuXH��ZhongL��HuangS��etal.Size-dependentfractureofsiliconnanoparticlesduringlithiation[J].ACSNano��2012��6(2)��1522-1531.